Find me in: Researchgate Orcid Academia Google Scholar
Our Research Group: Multiscale Mechanical Modeling and Research Network (MMMRN)
Computational Mechanics
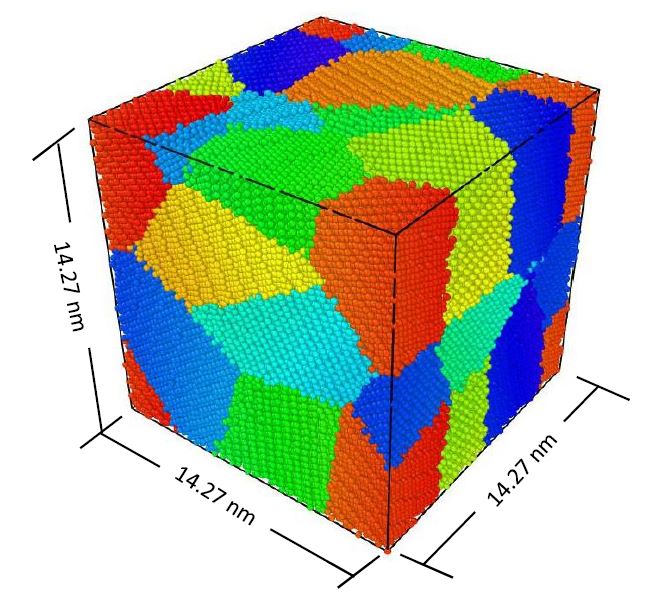
Mechanical Properties of Polycrystalline Tungsten Nanowire
Metal nanowires are stirring the attention of scientific world because of their many unique properties. W single crystal nanowires, with bcc crystal configuration, have become focus of many studies due to lack of complete insight of their mechanical deformation. However, polycrystalline W nanowires are not thoroughly studied yet. In this work, molecular dynamics simulation is used to identify different aspects of deformation and plasticity of polycrystalline W nanowire using EAM potential and a constant strain rate of 10 to the power 9 second inverse one. Impact of grain size, diameter, and temperature on the elastic properties is analyzed. Average grain size is varied from 4.63 nm to 25 nm keeping the diameter constant at 5 nm and temperature at 10 K. Diameter of the nanowire is varied from 2 to 5 nm keeping the average grain size and length to diameter ratio constant while temperature was changed from 10 K to 500 K maintaining fixed diameter and grain size. It is observed that inverse Hall-Petch behavior dominates the plasticity in polycrystalline nanowires and grain boundary sliding becomes the dominating mechanism of plasticity with a critical grain size. Nanowires with shorter diameter are found to be stronger and elevated temperature weakens the nanowire irrespective of the grain size.
.jpg)

Stress, grain size, and temperature dependent creep in Tungsten at Nanoscale
Tungsten, a transition metal with body-centered cubic crystal structure, has unique properties that enables its use in high temperature environment such as key material in filaments. At high temperature and stress creep becomes a key mechanism of deformation of material. In this work, for the first time, nature of creep in nanocrystalline tungsten and factors that govern creep mechanism like grain size, temperature, and applied stress are studied through atomistic simulation. Applied stress is varied from 2.5 to 5.5 GPa on the metallic sample while temperature is varied in the range of 1600 K - 2200 K. Stress and temperature variance are repeated for average grain sizes of 2.38 nm, 2.86 nm, 3.57 nm, and 4.76 nm. From simulation it has been found that the creep mechanism in nanocrystalline tungsten in contingent on the applied stress as the creep mechanism varies from grain boundary diffusion creep to dislocation dominated creep. Moreover, temperature and grain refinement seems to aid the creep phenomenon at nanoscale. In order to quantify the propensity and mechanism of creep in nanocrystalline tungsten stress and grain size exponents are observed in addition to the time evolution of stress. Finally, atomistic features of deformation are analyzed to evince the simulation results.
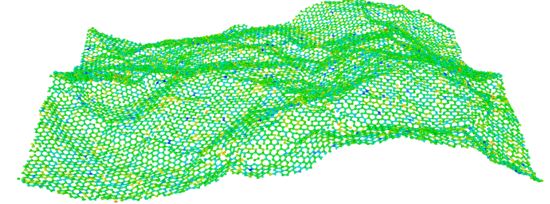
Graphene and its elemental analogue: A molecular dynamics view of fracture phenomenon
This study revealed the fracture response of graphene, hBN and silicene nanosheets under different tiny crack lengths by molecular dynamics (MD) simulations using LAMMPS. Our study shows a comparative analysis of mechanical properties among the elemental analogues of graphene and suggested that hBN can be a good substitute for graphene in terms of mechanical properties. We have also found that the pre-cracked sheets fail in brittle manner and their failure is governed by the strength of the atomic bonds at the crack tip. The MD prediction of fracture toughness shows significant difference with the fracture toughness determined by Griffth's theory of brittle failure which restricts the applicability of Griffith's criterion for these materials in case of nano-cracks. Moreover, the strengths measured in armchair and zigzag directions of nanosheets of these materials implied that the bonds in armchair direction have the stronger capability to resist crack propagation compared to zigzag direction.
Atomistic Representation of Anomalies in the Failure Behaviour of Nanocrystalline Silicene
Collaborator: Dr. Mahububul Islam, Purdue University
Silicene, a 2D analogue of graphene, has spurred a tremendous research interest in the scientific community for its unique properties essential for next generation electronic devices. In this work, for the first time, we present a molecular dynamics (MD) investigation to determine the fracture strength and toughness of nanocrystalline silicene (nc silicene) sheet of varied grain size and pre existing crack length at room temperature. Our results suggest that the transition from an inverse pseudo Hall-Petch to a pseudo Hall-Petch behavior in nc silicene occurs at a critical grain size of 17.32 nm. This phenomenon is also prevalent in nanocrystalline graphene. However, nc silicene with pre existing cracks exhibits anomalous crack propagation and fracture toughness behaviour. We have observed two distinct types of failure mechanisms (crack sensitive and insensitive failure) and devised the mechanophysical conditions under which they occur. Fracture toughness calculated from both Griffiths theory and MD simulations indicate that the former overpredicts the fracture toughness of nc silicene. The most striking outcome, however, is that despite the presence of a pre existing crack, the crack sensitivity of nc silicene is found to be dependent on the grain size and their orientations. This study is the first direct comparison of atomistic simulations to the continuum theories to predict the anomalous behaviour in deformation and failure mechanisms of nc silicene.
Computational Heat Transfer
My undergraduate research was fully focused on computational heat transfer especially convection heat transfer inside cavities of different geometries. It involved thermo-fluid modeling using FEM. Currently, I am working on this topic in collaboration with Dr. Suvash Saha and Dr. M. M. Rahman.